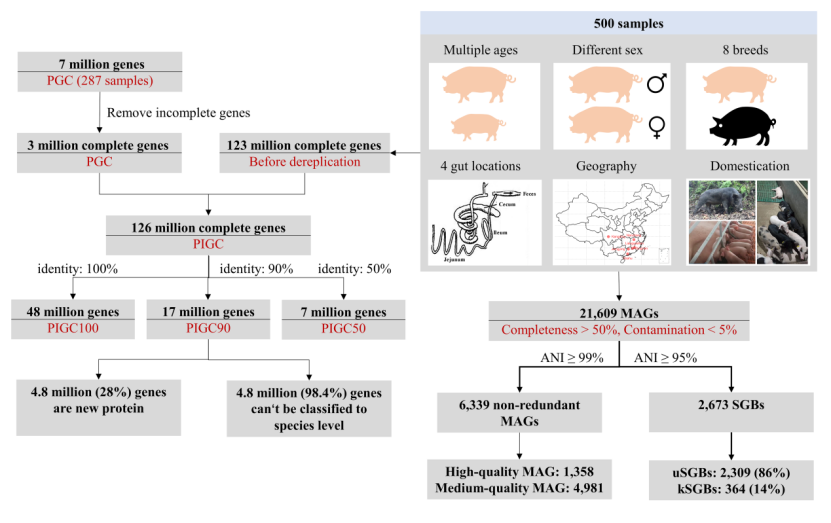
2月17日,黄路生院士团队在《Nature Communications》发表重大科研成果,提供了目前为止规模最为宏大的猪肠道微生物基因集和基于宏基因组组装的微生物基因组。黄路生院士及学校首席教授陈从英作为共同通讯作者、2018级博士研究生周云燕为共同第一作者、beat365官方网站为唯一通讯作者单位,发表题为“Expanded catalog of microbial genes and metagenome-assembled genomes from the pig gut microbiome”的研究论文,该研究通过使用广泛来源的猪肠道菌群样本,通过深度宏基因组测序,构建了整合的猪肠道微生物基因目录(PIGC),其中包含来自787个肠道元基因组的17,237,052个完整基因,它们以90%的蛋白质同一性聚类, 其中28%是未知蛋白质。
黄路生院士团队使用分箱分析,构建了6339个基于宏基因组组装的微生物基因组(MAG),它们被聚集成2673个物种级别的基因组仓(SGB),其中超过86%(2309)在当前数据库中没有可用的基因组序列(未知SGB,uSGB)。使用构建的PIGC和MAG,该研究比较了野猪和高度商业化杜洛克猪肠道微生物组之间的差异。发现两者之间肠道菌群组成差异显著,为从肠道菌群角度分析野猪抗逆性和商业猪种高生长速度、高饲料利用提供初步参考数据。同时,PIGC和MAG为猪肠道微生物组相关研究提供了扩展的资源。
家猪提供了人类食用的大部分肉,同时还是生物医学研究的动物模型。猪的胃肠道中有数万亿细菌,它们在宿主的新陈代谢、免疫甚至行为中起着至关重要的作用。在肠道菌群与猪重要经济性状的因果关系研究中依赖于微生物基因组的可用注释信息。在目前的数据库中,只有部分微生物的基因组及相关功能基因的注释信息。参考基因以及高质量的微生物基因组是了解特定微生物的功能作用并量化其在肠道微生物组中的丰度的必不可少的资源。但是,约40–50%的肠道微生物缺乏参考基因组。
与易受偏倚,敏感性低以及肠道微生物组缺乏功能性信息的16S rRNA基因测序相比,宏基因组测序可用于推断微生物群落的生物学功能,并已逐渐用于基于宏基因组关联分析挖掘肠道微生物组与宿主表型之间关联的相关研究中。但使用目前常用的组装方法进行宏基因组测序数据分析时,可能会产生错配和嵌合重叠群,并将明显的偏倚引入结果。因此,迫切需要完整的微生物基因目录和参考基因组序列信息来研究肠道微生物组。
在这项工作中,黄路生院士及学校首席教授陈从英带领团队通过对来自不同年龄、性别、品种、地理区域、不同肠道部位和野猪来源的500个样本,进行深度宏基因组测序,并整合了现有的猪肠道菌群基因组,构建了一个目前为止较为完整的猪肠道微生物基因目录和基于宏基因组组装的基因组。尤其是野猪肠道菌群样品以及空肠、回肠和盲肠内容物样品的使用,增强了PIGC和MAG的广泛代表性。
作为构建的猪肠道微生物基因集和MAG使用的范例,该研究还比较了野猪和高度商业化杜洛克两种代表了截然不同的饲养条件下猪肠道微生物组。PIGC和MAG为猪肠道微生物组相关研究提供了重要的资源。
https://www.nature.com/articles/s41467-021-21295-0
转载自:学校官网
